 2016, Vol. 47
2016, Vol. 47引用本文 | doi: 10.7501/j.issn.0253-2670.2016.23.005 |



2. 西南林业大学 西南山地森林资源保育与利用重点实验室, 云南 昆明 650224
2. Key Laboratory for Forest Resources Conservation and Use in the Southwest Mountains of China, Southwest Forestry University, Kunming 650224, China
小远志Polygala sibirica L. var. megalopha Fr.是云南一种民间药用植物,主要用于解热散寒和祛风止痛[1]。文献报道其中主要成分为黄酮类化合物[2-3]。本课题组前期研究工作中将小远志75%乙醇提取物通过D101大孔树脂柱色谱,依次用水和35%、65%、95%乙醇洗脱,并采用尿酸生成法来测定4部分洗脱物对黄嘌呤氧化酶(XO)的抑制活性。表 1显示65%乙醇洗脱物有更高的抗XO的活性。当样品质量浓度达到10.5 μg/mL时,其抑制率超过了50%,而其他的洗脱物活性稍低。同时,当样品质量浓度降低至1.75 μg/mL时,其余提取物抑制率已经不显著,而65%醇洗脱物仍然具有31.6%的抑制率。因此,本实验对体外XO抑制活性较高的小远志65%乙醇洗脱物进行化学成分的分离,以期发现其中对XO抑制率更高的单体化合物。通过分离纯化,结合活性追踪,得到4个对XO有较高抑制活性的糖酯类化合物,分别鉴定为远志寡糖酯A(tenuifoliside A,1)、3, 6′-二芥子酰基蔗糖(3, 6′-disinapoyl sucrose,2)、3′-E-3, 4, 5-三甲氧基肉桂酰基-6-苯基蔗糖(3′-E-3, 4, 5-trimethoxycinnamoyl-6-benzoyl sucrose,3)、3′-E-3, 4, 5-三甲氧基肉桂酰基-4-苯基蔗糖(3′-E-3, 4, 5-trimethoxycinnamoyl-4-benzoyl sucrose,4),均为首次从该植物中分离得到。
|
|
表 1 小远志各洗脱部分体外抗XO活性(x±s, n=3) Table 1 In vitro inhibitory activity of elution parts against XO (x±s, n=3) |
糖酯类物质具有一定的生物活性,如抗菌[4]、免疫调节[5]等,但是对于抗XO的活性未见报道。XO高效抑制剂有助于减少尿酸的生成,能够缓解和治疗痛风症状[6]。本实验分离得到的糖酯类化合物与对照品别嘌呤醇相比较,对XO有更好的抑制活性,同时作为天然来源的化合物,避免了别嘌呤醇严重的副作用[7],为进一步开发新的抗痛风药物奠定了基础。
1 仪器与材料Bruker AM-500兆超导核磁共振仪(Bruker公司);Bruker HTC/Esquire spectrometer型LC-MS(Bruker公司);Agilent 1100高效液相色谱仪(Agilent公司);半制备色谱柱为ZORBAX SB-C18反相柱(Agilent公司);UV-2450紫外可见分光光度计(北京岛津仪器有限公司)。薄层色谱硅胶板GF254板和柱色谱硅胶为青岛海洋化工厂产品;凝胶Sephadex LH-20(Amershm生科公司);反相填充材料RP18(40~60 μm,Merck公司)。XO(批号1001669570)溶于牛奶中,购于西格玛奥德里奇(上海)贸易有限公司;别嘌呤醇(批号HFBM160120319003),黄嘌呤(批号HX0429)购于合肥博美生物有限公司。
小远志Polygala sibirica L. var. megalopha Fr.干燥全草5 kg,2012年8月采自云南省昭通市永善县。由西南林业大学杜凡教授鉴定,标本(2012010)保存在西南林业大学林学院植物资源利用系药用植物教研室。
2 提取与分离小远志干燥全草约5 kg,粉碎后用75%乙醇回流提取3次,合并提取液,减压浓缩回收乙醇,得到浸膏1.7 kg。浸膏用温水溶解后,经D101大孔吸附树脂柱色谱,依次用水和35%、65%及95%乙醇洗脱。65%乙醇洗脱液回收溶剂后得到480 g粗提样品。样品经硅胶柱色谱,用氯仿-甲醇(1:0→0:1)梯度洗脱,得到了7个组分Fr. 1(31 g)、2(56 g)、3(47 g)、4(57 g)、5(122 g)、6(13 g)、7(106 g)。Fr. 2经硅胶柱色谱(氯仿-甲醇9:1→0:1)梯度洗脱,得到3个组分A、B和C。B(40 g)经RP18(甲醇-水30:70→70:30)洗脱后得到11个部分。B4部分(0.2 g)经过硅胶柱色谱(氯仿-甲醇20:1→1:1)洗脱得到12个部分,B4-7经Sephadex LH-20(氯仿-甲醇1:1)除掉部分色素后,再经过半制备型HPLC分离,乙腈-水(47:53)洗脱,得到化合物1(126 mg)、2(25 mg)。Fr. 4经过硅胶柱色谱(氯仿-甲醇10:1→0:1)洗脱得到10个部分(A~J)。E部分(10 g)经过RP18(甲醇-水20:80→70:30)洗脱后,得到14个部分。E1(161 mg)经半制备型HPLC分离,乙腈-水(10:90)洗脱,依次得到化合物3(25 mg)、4(17 mg)。化合物结构见图 1。

|
图 1 化合物1~4的结构 Fig.1 Structures of compounds 1-4 |
3 结构鉴定
化合物1:白色无定形粉末,根据负离子ESI-MS谱给出准分子离子峰m/z: 681 [M-H]-,结合13C-DEPT NMR谱,推测分子式为C31H38O17,不饱和度为13。1H-NMR (500 MHz, Pyridine-d5) δ: 7.89 (1H, d, J=8.4 Hz, H-3′′′, 5′′′), 7.71 (1H, d, J=15.9 Hz, H-7″), 6.54 (1H, d, J=15.9 Hz, H-8″), 6.95 (2H, s, H-2″, 6″), 6.81 (1H, d, J=8.4 Hz, H-2′′′, 6′′′), 5.49 (1H, d, J=3.6 Hz, H-1), 3.86 (6H, s, OMe), 3.78 (3H, s, OMe);13C-NMR (125 MHz, Pyridine-d5) δ: 166.7 (C-7′′′), 166.3 (C-9″), 163.5 (C-4′′′), 154.0 (C-3″, 5″), 145.7 (C-7″), 140.7 (C-4″), 132.4 (C-2′′′, 6′′′), 130.3 (C-1″), 121.4 (C-1′′′), 117.6 (C-8″), 116.0 (C-3′′′, 5′′′), 106.2 (C-2″, 6″), 104.7 (C-2′), 93.0 (C-1), 84.6 (C-5′), 79.8 (C-3′), 75.0 (C-3), 73.6 (C-4′), 73.1 (C-2), 72.3 (C-5), 71.1 (C-4), 65.6 (C-1′), 64.5 (C-6), 62.7 (C-6′), 56.1, 60.5 (OMe)。以上光谱数据与文献报道[4]基本一致,故鉴定化合物1为远志寡糖酯A。
化合物2:白色无定形粉末,根据负离子ESI-MS谱给出准分子离子峰m/z: 753 [M-H]-,结合13C-DEPT NMR谱,推测分子式为C34H42O19,不饱和度为14。1H-NMR (500 MHz, Pyridine-d5) δ: 7.70 (1H, d, J=15.9 Hz, H-7″), 7.63 (1H, d, J=15.9 Hz, H-7′′′), 7.05 (2H, s, H-2″, 6″), 7.03 (4H, s, H-2′′′, 6′′′), 6.54 (1H, d, J=15.9 Hz, H-8″), 6.45 (1H, d, J=15.9 Hz, H-8′′′), 5.48 (1H, d, J=3.3 Hz, H-1), 3.87 (6H, s, OMe), 3.85 (6H, s, OMe);13C-NMR (125 MHz, Pyridine-d5) δ: 167.6 (C-9″), 166.7 (C-9′′′), 149.1 (C-3″, 3′′′, 5″, 5′′′), 146.5 (C-7″), 145.9 (C-7′′′), 140.4 (C-4″, 4′′′), 125.1 (C-1″), 125.0 (C-1′′′), 115.4 (C-8″), 115.1 (C-8′′′), 106.8 (C-2″, 6″), 106.6 (C-2′′′, 6′′′), 104.7 (C-2′), 92.7 (C-1), 84.7 (C-5′), 79.3 (C-3′), 75.2 (C-3), 74.0 (C-2), 73.1 (C-4′), 72.2 (C-5), 71.6 (C-4), 65.7 (C-1′), 65.1 (C-6), 63.3 (C-6′), 56.2, 56.3 (OMe)。以上数据与文献报道[4]基本一致,故鉴定化合物2为3, 6′-二芥子酰基蔗糖。
化合物3:白色无定形粉末,根据正离子ESI-MS谱给出准分子离子峰m/z: 689 [M+Na]+,结合13C-DEPT NMR谱,推测分子式为C31H38O16,不饱和度为13。1H-NMR (500 MHz, CD3OD) δ: 7.71 (1H, d, J=15.9 Hz, H-7″), 6.54 (1H, d, J=15.9 Hz, H-8″), 6.95 (2H, s, H-2″, 6″), 5.48 (1H, d, J=3.6 Hz, H-1), 3.86 (6H, s, OMe), 3.78 (3H, s, OMe);13C-NMR (125 MHz, CD3OD) δ: 168.0 (C-7′′′), 167.7 (C-9″), 154.8 (C-3″, 5″), 147.2 (C-7″), 141.2 (C-4″), 134.3 (C-4′′′), 131.4 (C-1″), 131.2 (C-1′′′), 130.7 (C-3′′′, 5′′′), 129.6 (C-2′′′, 6′′′), 117.7 (C-8″), 106.8 (C-2″, 6″), 104.8 (C-2′), 93.1 (C-1), 84.1 (C-5′), 79.6 (C-3′), 74.9 (C-3), 74.0 (C-4′), 73.0 (C-2), 72.4 (C-5), 71.5 (C-4), 65.5 (C-1′), 65.5 (C-6), 63.4 (C-6′), 56.6, 61.1 (OMe)。以上数据与文献报道[8]基本一致,故鉴定化合物3为3′-E-3, 4, 5-三甲氧基肉桂酰基-6-苯基蔗糖。
化合物4:白色无定形粉末,根据正离子ESI-MS谱给出准分子离子峰m/z: 689 [M+Na]+,结合13C-DEPT NMR谱,推测分子式为C31H38O16,不饱和度为13。1H-NMR (500 MHz, CD3OD) δ: 7.72 (1H, d, J=15.9 Hz, H-7″), 6.54 (1H, d, J=15.9 Hz, H-8″), 6.93 (2H, s, H-2″, 6″), 5.47 (1H, d, J=3.6 Hz, H-1), 3.86 (6H, s, OMe), 3.78 (3H, s, OMe);13C-NMR (125 MHz, CD3OD) δ: 168.0 (C-7′′′), 167.8 (C-9″), 154.8 (C-3″, 5″), 147.2 (C-7″), 141.2 (C-4″), 134.3 (C-4′′′), 131.5 (C-1″), 131.3 (C-1′′′), 130.7 (C-3′′′, 5′′′), 129.6 (C-2′′′, 6′′′), 117.8 (C-8″), 106.8 (C-2″, 6″), 104.9 (C-2′), 93.1 (C-1), 84.1 (C-5′), 79.6 (C-3′), 74.9 (C-3), 74.0 (C-4′), 73.2 (C-2), 72.4 (C-5), 76.6 (C-4), 65.6 (C-1′), 60.8 (C-6), 63.4 (C-6′), 56.7, 61.2 (OMe)。以上数据与文献报道[8]基本一致,故鉴定化合物4为3′-E-3, 4, 5-三甲氧基肉桂酰基-4-苯基蔗糖。
4 抗XO活性测试抗XO活性实验参考文献方法[9]进行改进。按照表 2顺序依次加入不同试样(其中供试样浓度分别预先配制成1.75、3.50、7.00、10.50 μg/mL),混合均匀,水浴温度为25 ℃,反应时间30 min,用紫外分光光度计测定各处理样品,在290 nm处检测吸光度(A)值。根据以下公式计算不同质量浓度样品对XO的抑制率(表 3)。其中,A1~A4为实验处理1~4在290 nm处的A值。
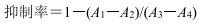
|
|
|
表 2 抗XO试验反应体系 Table 2 Reaction system of XO inhibiting test |
|
|
表 3 糖酯与对照品别嘌呤醇体外抗XO活性(x±s, n=3) Table 3 In vitro inhibitory activity of glycolipids and allopurinol against XO (x±s, n=3) |
表 3显示了4个糖酯与对照品别嘌呤醇体外抗XO活性。化合物1、3、4的抑制活性(IC50分别为9.5、10.2、7.7 μmol/L)比对照品别嘌呤醇(IC50=11.2 μmol/L)略高,而化合物2(IC50=1.3 μmol/L)则显示了比对照品更高的抑制活性。结合结构,推测其中的苯乙烯基与糖基相连的片段在空间上与XO有较高的适应性,可以与黄嘌呤竞争,从而起到抑制尿酸生成的作用。化合物2相比其他3个糖酯类化合物,可能由于分子中含有2个起效片段,其抑制效果更强。
5 结论本实验根据活性追踪,分离得到了4个对XO有较强抑制活性的糖酯类化合物,并初步对构效关系提出了一些假设,为进一步研究构效关系提供了方向。这一类新型的高效抗XO的化合物,也有着进一步被开发成治疗痛风药物的潜力。
| [1] | 中国科学院昆明植物研究所. 中国植物志[M]. 北京: 科学出版社, 1997 . |
| [2] | 宛瑜, 郁慧珍, 叶玲. 从小远志中分离黄酮醇和口山酮衍生物[J]. 西南大学学报:自然科学版 , 2014, 30 (7) :51–55. |
| [3] | Huang Y J, Zhou L Y, Wang J M, et al. Two new flavonol glycosides from Polygala sibirica L. var. megalopha Fr.[J]. Molecules , 2015, 20 (12) :21494–21500. DOI:10.3390/molecules201219775 |
| [4] | Jiang Y, Tu P F, Chen X J. Isolation of two sucrose esters from Polygala tenuifolia by high speed countercurrent chromatography[J]. J Liq Chromatogr Relat Technol , 2005, 28 (10) :1583–1592. DOI:10.1081/JLC-200058371 |
| [5] | Zou M, Yu S S, Wang K, et al. Glycosylation of ent-kaurene derivatives and an evaluation of their cytotoxic activities[J]. Chin J Nat Med , 2013, 11 (3) :289–295. |
| [6] | 韩莹, 朱翊, 傅得兴. 非布司他治疗高尿酸血症伴痛风的药理与临床评价[J]. 中国新药与临床杂志 , 2010, 29 (8) :635–638. |
| [7] | Umamaheswari M, Asokkumar K, Somasundaram A, et al. Xanthine oxidase inhibitory activity of some Indian medical plants[J]. J Ethnopharmacol , 2007, 109 (3) :547–511. DOI:10.1016/j.jep.2006.08.020 |
| [8] | Miyase T, Ueno A. Sucrose derivatives from the roots of Polygala tenuifolia[J]. Shoya Kugaku Zashi , 1993, 47 (3) :267–276. |
| [9] | 齐万虎, 蒋企洲, 蒋建勤. 黄瑞香中化学成分抑制黄嘌呤氧化酶活性及其抑制机制[J]. 中国实验方剂学杂志 , 2014, 20 (5) :141–144. |