 2014, Vol. 45
2014, Vol. 45引用本文 | doi: 10.7501/j.issn.0253-2670.2014.10.014 |



2. 天津药物研究院 天津市新药设计与发现重点实验室, 天津 300193
2. Tianjin Key Laboratory of Molecular Design and Drug Discovery, Tianjin Institute of Pharmaceutical Research, Tianjin 300193, China
血小板能够被凝血酶、ADP、血栓素A和胶原等激活剂激活[1],在这些激活剂中,凝血酶是最强的血小板聚集激活剂。凝血酶激活血小板聚集是通过凝血酶蛋白水解细胞表面蛋白酶激活受体(protease activated receptor,PAR)来实现的,PAR有多个亚型,其中PAR-1是这些亚型中最主要的受体,可作为黑色素瘤治疗的潜在靶点[2]。PAR-1拮抗剂能阻断凝血酶激活血小板,从而抑制血小板聚集和血栓形成,PAR-1是一个前景良好的全新抗血小板血栓的靶标,目前有SCH 530348(Vorapaxar)和E5555(Atopaxar)处在III期和II期临床研究阶段[3]。本实验从虚拟筛选和实验筛选2方面对30个中药成分进行考察,寻找潜在的PAR-1拮抗剂。
1 材料 1.1 药品与试剂凝血酶受体激动肽(TRAP),生工生物工程(上海)有限公司,生产批号061112;枸橼酸钠,国药集团化学试剂有限公司,生产批号20120109。E5555,自制,质量分数均≥95%,批号20110109,30种植物成分,自制,质量分数均≥95%。
1.2 动物SPF级雄性豚鼠,体质量(450±20)g,购自天津山川红实验动物有限公司,合格证号为SCXK(津)2011-0001。
1.3 仪器LBY-NJ四通道血小板聚集仪,北京普利生仪器有限公司。
2 方法 2.1 植物成分的虚拟筛选 2.1.1 配体准备30个植物成分(T1~T30),见表 1,阳性药SCH 530348和E5555。应用Schrődinger准备结构,以Ligprep对小分子配体进行处理,保持立体结构与天然一致。
|
|
表 1 30个植物成分的编号及名称 Table 1 Codes and names of 30 ingredients from plants |
在RCSB 蛋白数据库(http://www.rcsb.org)下载人源PAR-1与配体SCH530348的复合物三维晶体结构(代码3VW7)[4]。用Schrődinger软件对受体分子进行处理,给蛋白加氢并将晶体结构中的水分子和杂分子除去。以原配体SCH 530348为中心,设置盒子大小设为1.2 mm,其他参数均为默认值,生成格点文件备用。
2.1.3 分子对接利用Schrődinger软件中的Glide模块进行对接计算,其过程是寻找配体与受体在活性位点结合处结合能较低的构象,对接参数均为默认值,记录配体与对接位置各残基的氢键相互作用。
2.2 植物成分体外拮抗PAR-1活性的筛选豚鼠颈动脉采血,以3.8%柠檬酸钠(9∶1)混合抗凝,室温下离心分离(200 g×10 min),得混合富血小板血浆(PRP),剩余部分再以1 000×g离心10 min,取上清液为贫血小板血浆(PPP),用PPP调PRP血小板计数为2×105~6×105 / μL,按Born比浊法测定血小板聚集程度。用PPP调零(100%),在比浊管中加入267 μL PRP,同时加入30 μL待测化合物,使其终浓度为1 μmol/L,搅拌1 min后,加入3 μL刺激剂TRAP(终浓度为20 μmol/L),记录3 min内血小板最大聚集率,以E5555作为阳性对照,生理盐水作为空白对照。每个实验组以3次平行测定所得均值进行统计学处理,按公式计算药物的抑制率。
抑制率=(空白对照组聚集率-给药组聚集率)/ 空白对照组聚集率
2.3 数据处理
对接得分、结合能直接采用计算数据。空间结合模式根据对接后配体位置,以及结合位置的空腔分布来定性判定。氢键结合情况是根据对接结果中记录的配体与对接位置各残基的氢键相互作用进行分级,分值在-0.3~-0.6为弱氢键,在-0.7~-1.3为中等氢键、-1.3~-2为强氢键。体外筛选实验所得数据以 ±s表示。
±s表示。
530348与PAR-1的结合模式分析 PAR-1是一个GPCR受体,有7次跨膜,图 1是PAR-1与SCH 530348晶体复合物的结构,显示了其跨膜区及胞内区,SCH 530348结合在胞外区。进一步放大局部结构,得到图 2,显示了SCH 530348的PAR-1结合位点周围的残基及与其形成的3个氢键(Leu258、Tyr337、Ala349)。从残基性质来看,包含酸性残基GLY233和ASP256,带羟基的残基Tyr183、Tyr187、Tyr337、Thr343、Ser344、Tyr350、
 | 图 1 PAR-1与SCH 530348晶体复合物立体结构 Fig. 1 Cocrystalline structure of PAR-1 and SCH 530348 compound |
 | 图 2 SCH 530348与PAR-1结合位点周围的残基及形成的氢键 Fig. 2 Residues and hydrogen bonds around binding site of SCH 530348 and PAR-1 |
Tyr353,带咪唑的残基His255、His336,其余全是疏水性氨基酸残基Val234、Pro236、Leu237、Val257、Leu258、Leu262、Leu263、Phe271、Leu330、Leu333、Leu340、Ala348、Ala349、Ala352。从空间上看,极性残基分布在图两侧,这个空腔总体比较适合非极性分子,空腔表面见图 3,呈类似T或Y型,为了便于进一步分析空腔的占据憔,将其分解为5部分,分别在图中表示为I、II、III、IV、V。
 | 图 3 SCH 530348的PAR-1结合位点空腔的形状及分解 Fig. 3 Shape and decomposition of cavity of binding site for SCH 530348 and PAR-1 |
SCH 530348的对接位置与晶体复合物中的位置完全重合,对接得分达到-14.9的高分,结合能为-336.2 kJ/mol。根据图 3可以描述其与PAR-1的结合模式为SCH 530348分子总体疏水性较强,占据了空腔的5个区域,为数不多的杂原子在II、III、IV区域与受体形成3个氢键。
3.1.2 阴性药与PAR-1的结合模式分析虚拟筛选与实验筛选一样,要正确评价虚拟活性,除了阳性对照,也必须要与阴性对照比较,排除噪音,得到有效信息。本研究采用盐酸氨溴索等7个分子结构作为阴性对照,从表 2对接得分上可以看出,3个分子在-9.0~-10.0,属于得分比较高的范围,其他4个分子在-6.1~-6.4,得分较低。从氢键来看,得分最高的2个分子形成2个以上较强的氢键。从表 3的配体占据空腔的模式来看,阴性配体出现在4、5、9等3种模式中。总体上分析,PAR-1的空腔具有较大的空间,大部分结构小的分子都能够对接进来。
结合阳性药和阴性药的情况,虚拟筛选的策略为应该考虑分子的疏水性、占据空腔的4部分区域以上,并且具有适当的氢键结合。
|
|
表 2 各配体与PAR-1对接得分、结合能和氢键强度 Table 2 Docking score,binding energy,and hydrogen bond strength between each ligand and PAR-1 |
|
|
表 3 配体占据PAR-1空腔的模式 Table 3 Occupied modes of ligands in cavity of PAR-1 |
为了寻找新型PAR-1拮抗剂,同时验证虚拟筛选的策略,对本实验室现有的30个植物成分进行了虚拟筛选,结果见表 2和3,其中T10、T11、T12 3个化合物对接后无结果,猜测是因物质立体结构与受体空腔位置不符。根据得分不低于阴性对照中位值(-9.0)的原则,T30、T6、T21、T24、T9等具有较大希望,进一步分析氢键T24和T9可以被排除。
从表 3的空间占据模式来看,T21与阳性对照SCH 530348具有类似的模式。根据占有4个区域以上的策略分析,T24和T30具有较大希望。由于T24没有氢键,从虚拟筛选总体策略来看,从理论上可以得到的推论是T30和T21具有PAR-1拮抗作用的前景。
3.2 植物成分体外拮抗PAR-1活性的筛选结果为了使筛选的结果更有成药的实用价值,本实验采用较低终浓度(1 μmol/L)进行30个化合物和阳性药E5555对抗20 μmol/L的TRAP诱导豚鼠血小板聚集有抑制作用的筛选剂,由于没有SCH 530348样品,实验中以E5555为阳性对照,抑制率达99.7%,抑制率小于15%的药物在本实验条件下作用强度较低,包括T1~11、T13~21、T23、T24、T26、T27、T30,抑制率超过15%药物见表 4。
|
|
表 4 化合物对TRAP诱导血小板聚集的抑制率 (±s,n=3)
Table 4 Inhibitory rates of compounds on TRAP-induced platelet aggregation (±s,n=3)
|
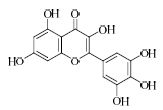
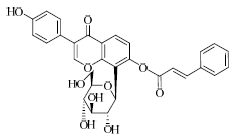
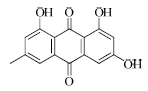
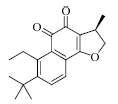
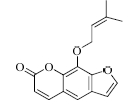
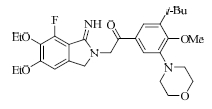
结果表明,具有比较明确的PAR-1体外拮抗作用的植物成分为T5、T21、T28、T29,T25具有一定的作用,其中T21抑制率最大并且与虚拟筛选的预期结果一致。与虚拟筛选结果预期差异较大的有二类,一类是理论上活性较好的T30实验中没有显示活性,另一类是虚拟筛选没有发现的T5、T28、T29,其活性接近T21。
对于T21的作用与虚拟筛选的预期一致,表明在深入研究SCH 530348与受体结合基础上,提出的虚拟筛选策略是成功的,结合模式见图 4。仔细分析比较T21与SCH 530348分子及其与PAR-1的结合模式,T21在分子的亲水性占较大优势,在空腔V区的占据较小,在IV、V区缺乏氢键支持,所以活性与阳性药相比还有一定差别,也说明了这些差异的重要性,在以后的改造和设计中要充分体现出来。
 | 图 4 T21 (A) 和T5 (B) 与PAR-1活性位点结合模式 Fig. 4 Binding modes of T21 (A) and T5 (B) with active site of PAR-1 |
T30在理论筛选上体现较好的活性预期,实验中没有显示活性,这种理论筛选的假阳性非常普遍,仔细分析T30与T21和阳性分子的差别可以发现,T30尽管对接得分好于T21,主要得益于与受体空腔的II、III、IV、V匹配较好,主要的不足是缺少I、II、III区与受体较强的氢键作用,由此可以推测:与受体残基258作用的氢键对于产生活性具有重要作用,相比而言,重要性超过IV区的氢键。
虚拟筛选没有发现的T5、T28、T29活性,是典型的假阴性现象,根据基于阳性药的模式制定虚拟策略是不可能被发现的。3个分子基本都是一种模式,对接得分与阴性对照无异,对空腔III区占据较满(图 4),只与残基258存在较强的氢键,这实际上可以理解为一种新的作用模式:仅凭与单一残基(258)较强作用和与单一区域(III区)匹配结合两条就可以具有较强的PAR-1拮抗活性,这也为寻找PAR-1拮抗剂的结构类别提供了新的方向。
4 讨论PAR是属于G蛋白偶联受体家族的一员,目前,主要有4种亚型,人的血小板表面只存在PAR-1和PAR-4,而介导血小板活化的凝血酶反应的主要受体是PAR-1,PAR-1受体拮抗剂可阻断凝血酶介导的血小板激活,而不会影响凝血酶介导的纤维蛋白原裂解,且PAR-1受体拮抗剂不影响参与血小板黏附、激活、聚集途径的相关因子,例如胶原,vWF,ADP和促凝血素,因此PAR-1拮抗剂在抗血栓的同时,可能具有出血风险低的特点[6]。现阶段处在临床研发中的PAR-1拮抗剂有SCH 530348和E5555 两种化合物[7,8],II期临床试验结果表明,E5555能够显著减少冠状动脉综合症心血管事件的发生并且没有增加出血风险[9,10]。因此,寻找PAR-1拮抗剂具有重要的意义。
本实验中,首先利用虚拟筛选,以SCH 530348做阳性对照,主要是根据SCH 530348与PAR-1的晶体结构进行分析,提出了框架性的虚拟筛选策略为:应该考虑分子的疏水性、占据空腔的4部分区域以上,并且具有适当的氢键结合。结合虚拟筛选和实验筛选的结果发现,T21抑制率最大,与虚拟筛选的预期一致,虚拟筛选假阳性的为T30,假阴性的有T5、T28、T29。通过对这5个分子与受体的对接情况的仔细分析,可以进一步明确虚拟筛选:残基258和空腔III对活性的影响是决定性的,其他各区在活性调节中II区主要是氢键和匹配的要求,IV和V区主要是疏水性匹配性,氢键结合对于进一步提升活性具有重要意义。
因PAR-1在灵长类动物和豚鼠血小板表面表达[11],采用豚鼠体外血小板对化合物进行筛选,本实验中采用的药物浓度为1 μmol/L的单浓度筛选,PAR-1受体激动肽浓度采用20 μmol/L,这结果主要体现的是体外靶标水平的竞争能力,从文献来看,初筛的浓度一般比较高,经常有10~20 μmol/L的报道,倾向于采用较低的浓度主要是使研究结果更接近成药实用价值。
本实验通过结合虚拟筛选和实验筛选开展研究,在常规的对接得分评价匹配性的基础上,增加了阴性对照、结合区域匹配和氢键相结合等细化评价内容,从预期的两个结构中,验证了一个活性较好结构,这种改进大大提高了成功率。通过对实验筛选假阴性分子与受体结合模式的深入分析,加深了对配体和受体相互作用的认识,这种理论与实验深度结合的研究模式是一种非常有意义的探索。
目前,还没有T21在抗血小板及血栓方面作用的报道,本实验发现将有助于其新的药效的开发研究。其他成分如杨梅素、大黄素、隐丹参酮、欧前胡素等在文献中都有抗血栓、抗心肌缺血、降低血液黏度或改善微循环等方面的作用报道[12,13,14],根据研究的结果,其作用机制可能与PAR-1拮抗作用相关,这也有利于阐明植物成分的作用机制。
| [1] | Davi G, Patrono C. Platelet activation and atherothrombosis [J]. N Engl J Med, 2007, 357(24): 2482-2494. |
| [2] | 周植星, 张 玥, 潘婷婷, 等. 凝血酶蛋白酶激活受体-1作为转移性黑色素瘤治疗潜在靶点的研究进展 [J]. 药物评价研究, 2012, 35(4): 276-280. |
| [3] | Kosoglou T, Reyderman L, Tiessen R G, et al. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorpaxar (formerly SCH 530348) in healthy subjects. [J]. Eur J Clin Pharmacol, 2012, 68(3): 249-258. |
| [4] | Zhang C, Srinivasan Y, Arlow D H, et al. High-resolution crystal structure of human protease-activated receptor 1 [J]. Nature, 2012, 492(7429): 387-392. |
| [5] | Coughlin S R. Protease-activated receptors in hemostasis, thrombosis and vascular biology [J]. J Thromb Haemost, 2005, 3(8): 1800-1814. |
| [6] | Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist SCH 530348 in acute coronary syndromes [J]. N Engl J Med, 2012, 366(1): 20-33. |
| [7] | Scirica B M, Bonaca M P, Braunwald E, et al. SCH 530348 for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2 degrees P-TIMI 50 trial [J]. Lancet, 2012, 380(9850): 1317-24. |
| [8] | Goto S, Ogawa H, Takeuchi M, et al. Double-blind, placebo-controlled Phase II studies of the protease-activated receptor 1 antagonist E5555 (atopaxar) in Japanese patients with acute coronary syndrome or high-risk coronary artery disease [J]. Eur Heart J, 2010, 31(21): 2601-2613. |
| [9] | O'Donoghue M L, Bhatt D L, Wiviott S D, et al. Safety and tolerability of atopaxar in the treatment of patients with acute coronary syndromes: the lessons from antagonizing the cellular effects of Thrombin-Acute Coronary Syndromes Trial [J]. Circulation, 2011, 123(17): 1843-1853. |
| [10] | Kogushi M, Matsuoka T, Kawata T, et al. The novel and orally active thrombin receptor antagonist E5555 (Atopaxar) inhibits arterial thrombosis without affecting bleeding time in guinea pigs [J]. Eur J Pharmacol, 2011, 657(1-3): 131-137. |
| [11] | 邱 麒, 郭素华, 臧宝霞, 等. 杨梅素对血小板活化因子拮抗的作用 [J]. 药学学报, 2003, 38(11): 831-833. |
| [12] | Zhou L M, Zuo Z, Moses S S, et al. Danshen: an overview of its chemistry, pharmacology, pharmaco- kinetics, and clinical use [J]. Clin Pharmacol, 2005, 45(12): 1345-1359. |
| [13] | Nie H, Meng L Z, Zhou J Y. Imperatorin is responsible for the vasodilatation activity of Angelica dahurica var. formosana regulated by nitric oxide in an endothelium-dependent manner [J]. Chin J Integr Med, 2009, 15(6): 442-447. |
| [14] | Zhang Y, Cao Y J, Zhan Y Z, et al. Furanocoumarins-im- peratorin inhibits myocardial hypertrophy both in vitro and in vivo [J]. Fitoterapia, 2010, 8(18): 1188-1195. |