 2017, Vol. 40
2017, Vol. 40引用本文 | doi: 10.7501/j.issn.1674-6376.2017.02.004 |



2. 天津药物研究院释药技术与药代动力学国家重点实验室, 天津 300193
2. State Key Laboratory of Drug Delivery Technology and Pharmacokinetics, Tianjin Institute of Pharmaceutical Research, Tianjin 300193, China
伊潘立酮(IPLT)是一种新型的非典型抗精神病药物,既是5-羟色胺2A(5-HT2A)受体阻滞药,又是多巴胺(D2)受体阻滞药,与目前使用的其他抗精神病药物相比,在有效治疗精神分裂症的同时可以减少锥体外系综合症的发生,安全性和耐受性良好。2009年5月,由美国Vanda制药公司开发的伊潘立酮片(商品名为Fanapt)获得了FDA的批准并在美国上市,在临床上用于治疗成年人的精神分裂症,共分为7种规格:1、2、4、6、8、10和12 mg[1]。伊潘立酮片的上市标志着对许多现有药物治疗仅部分有效的精神分裂症患者是一个新的机会,该药物可以更好地控制他们的症状。
多种pH值溶出曲线的测定已成为国内外评价固体制剂内在品质的一种重要科学手段,是提高仿制药生物等效性试验成功率的必经之路,本文以伊潘立酮为模型药物,进行了“pH-溶解度”曲线的测定,自研制剂与参比制剂的多溶媒溶出曲线的测定,并用f2相似性因子拟合,来探索仿制药体外溶出一致性的研究方法。
1 仪器与试药 1.1 仪器AL104天平(梅特勒-托利多仪器有限公司);RC12AD溶出试验仪(天津市天大天发科技有限公司);Waters e2695-e2489高效液相色谱仪(沃特世公司);FE20pH计(梅特勒-托利多仪器有限公司);TGL-16C离心机(上海安亭科学仪器厂)
1.2 药物与试剂伊潘立酮原料药(天津药物研究院化药部,批号R121217);伊潘立酮自研制剂(天津药物研究院制剂中心,规格1 mg/片,批号14091101);伊潘立酮参比制剂Fanapt(美国Vanda制药公司,规格1 mg/片,批号LOT FFZN);盐酸、氢氧化钠购自天津市凯信化学工业有限公司,分析纯;乙酸钠、磷酸二氢钾、无水柠檬酸、无水磷酸氢二钠、磷酸氢二铵购自天津市光复科技发展有限公司,分析纯;冰乙酸(天津市康科德科技有限公司,分析纯);甲醇(天津市康科德科技有限公司,色谱纯)
2 方法与结果 2.1 “pH-溶解度”曲线的测定 2.1.1 系列pH溶液的配制配制pH1.0~8.0系列溶液,盐酸溶液、pH4.5醋酸盐缓冲液、pH6.8磷酸盐缓冲液的配制参照《中国药典》2015年版[2],其他pH溶液采用McIlvaine缓冲液(用0.1 mol/L柠檬酸溶液和0.2 mol/L磷酸氢二钠溶液混合配制)和磷酸盐缓冲液(用0.2 mol/L磷酸二氢钾溶液和0.2 mol/L氢氧化钠溶液混合配制),测定各溶液pH。
2.1.2 “pH-溶解度”曲线的测定以上述pH1.0~8.0系列溶液500 mL作为溶出介质,采用桨法,转速为50 r/min,介质温度为(37±0.5)℃,每杯投入过量伊潘立酮原料药(经气流粉碎处理),使之形成过饱和溶液,经24 h后取样滤过,取续滤液,稀释,参照“溶出度试验测定方法”项下色谱条件,注入液相色谱仪,记录色谱图,计算伊潘立酮在不同pH溶液中的溶解度(S),结果见表 1。以pH为横坐标,以lgS值为纵坐标,绘制伊潘立酮“pH-溶解度”曲线,结果见图 1。
| 表 1 伊潘立酮在不同pH溶液中的溶解度(37 ℃) Table 1 Solubility of IPLT in different pH solutions (37 ℃) |

|
直线为最大规格剂量(12 mg)溶解在250 mL水中的溶解度 The straight line represents the solubility the maximum standard dose (12 mg) in the 250 mL water 图 1 pH-溶解度曲线(37 ℃) Fig. 1 pH Solubility profile of IPLT at 37 ℃ |
结果表明,伊潘立酮为难溶性药物,几乎不溶于水和pH6.8磷酸盐缓冲液,极微溶于0.1 mol/L盐酸溶液,微溶于pH4.5醋酸盐缓冲液。
2.1.3 不同pH溶液中稳定性的测定上述过饱和溶液,分别经24、48、72 h后取样滤过,取续滤液,稀释,分别注入液相色谱仪,记录色谱图,计算溶解度。结果表明,在(37±0.5)℃条件下,伊潘立酮在上述12个pH溶液中72 h内,溶解度RSD依次为0.41%、0.88%、0.28%、0.26%、0.89%、0.78%、1.80%、1.71%、1.54%、0.86%、1.91%、1.86%,均小于2.0%,未发生降解,稳定性良好。
2.2 溶出度测定方法 2.2.1 溶出介质的选择参照《普通口服固体制剂溶出度试验技术指导原则》[3]及伊潘立酮“pH-溶解度”曲线测定结果,选择4种不同pH溶液作为溶出介质:0.1 mol/L盐酸溶液、pH 4.5醋酸盐缓冲液、pH 6.8磷酸盐缓冲液、水。
2.2.2 溶出度测定方法(1)高效液相色谱条件 照高效液相色谱法(《中国药典》2015年版四部通则0512)进行测定。色谱柱为Thermo BDS HYPERSIL C18(250 mm×4.6 mm,5 μm);流动相为甲醇-0.01 mol/L磷酸氢二铵(磷酸调节pH至7.0±0.05)(70:30);检测波长229 nm;体积流量1.0 mL/min;柱温35 ℃;进样体积100 μL;运行时间10 min。
(2)专属性 分别取流动相、4种溶出介质、混合辅料、伊潘立酮对照品溶液、伊潘立酮片样品溶液各100 μL,分别注入液相色谱仪,记录色谱图,结果见表 2。结果表明,在该色谱条件下,流动相、溶出介质、混合辅料峰保留时间<4.4 min,伊潘立酮峰与各峰分离度良好,不干扰伊潘立酮片溶出度测定。
| 表 2 伊潘立酮片溶出度测定方法学专属性结果 Table 2 Specificity results of IPLT Tablets resolution method |
(3)线性范围 精密称取伊潘立酮对照品约10 mg,置100 mL量瓶中,加流动相适量使溶解,用流动相稀释至刻度,摇匀,分别精密移取0.5、1.0、1.5、2.0、2.5、3.0 mL置100 mL量瓶中,分别用4种溶出介质稀释至刻度,摇匀,注入液相色谱仪,以峰面积为纵坐标,质量浓度为横坐标,绘制标准曲线,计算回归方程。结果表明,在0~3.057 μg/mL浓度范围内,伊潘立酮在0.1 mol/L盐酸溶液、pH4.5醋酸盐缓冲液、pH6.8磷酸盐缓冲液、水中的线性回归方程及相关系数分别为A=293 493 C -2 835.5,r=0.999 8;A=284 709 C -3235.6,r=0.999 9;A=301 277 C -2 453.4,r=0.999 8;A=294 684 C -4048.9,r=0.999 8。伊潘立酮在4种溶出介质中线性关系均良好。
(4)回收率 精密称取伊潘立酮对照品适量,置量瓶中,按处方量加入混合辅料,分别用4种溶出介质溶解,稀释,配制成质量浓度为1.4、2.0、2.6 μg/mL的样品,每个质量浓度平行操作3份,滤过,注入液相色谱仪,记录色谱图,计算回收率。结果表明,伊潘立酮在0.1 mol/L盐酸溶液、pH4.5醋酸盐缓冲液、pH6.8磷酸盐缓冲液、水中的平均回收率分别为99.95%、99.62%、99.91%、99.74%;RSD分别为0.21%、0.65%、0.43%、0.78%。伊潘立酮在4种溶出介质中回收率均良好(n=9)。
(5)滤膜干扰 取伊潘立酮片1片,置500 mL溶出介质中,37 ℃水浴中振摇30 min,取出,分别经离心(11 000 r/min、7 min)、一次性针式过滤器(尼龙0.45 mm×15 mm)滤过,取上述溶液分别注入液相色谱仪,记录色谱图,考察滤膜干扰情况。结果表明,使用0.45 mm微孔滤膜滤过,弃初滤液5 mL,测定结果与离心测定结果一致,滤膜无吸附,不干扰测定;全溶出测定中滤液40 mL,滤膜无吸附,结果表明,使用0.45 mm微孔滤膜滤过不干扰伊潘立酮片溶出度的测定。
(6)稳定性 将伊潘立酮对照品溶液、样品溶液样品仓内(25±5)℃放置,于2、4、6、8、10、12、24、48 h取样100 mL,分别注入液相色谱仪,记录色谱图。
将对照品溶液、样品溶液置于4 ℃冰箱内冷藏放置,于1、2、3、4 d取样100 mL,分别注入液相色谱仪,记录色谱图。
结果表明,将对照品溶液、样品溶液于样品仓内放置48 h,峰面积RSD分别为0.86%、0.69%;将对照品溶液、样品溶液于4 ℃冰箱放置4 d,峰面积RSD分别为1.43%、1.98%,溶液稳定性良好。
(7)耐用性 改变高效液相色谱条件,考察色谱系统参数变化耐用性,包括:体积流量±0.1 mL/min、柱温±2 ℃,波长±2 nm、流动相比例±2%、流动相pH±0.2。设置高效液相色谱系统参数,分别取对照品溶液和供试品溶液注入液相色谱仪,记录色谱图,计算含量。结果表明各微调条件下,伊潘立酮测定结果的RSD值均小于1.0%(n=6),色谱系统参数耐用性良好。
2.2.3 溶出方法与数据处理照溶出度测定法(《中国药典》2015年版四部通则0931第二法),用以上4种溶媒500 mL作为溶出介质,温度(37±0.5)℃,转速50 r/min,取自研制剂和参比制剂各12片,依法操作,分别在5、10、15、20、30、45、60 min取溶液7 mL,0.45 μm微孔滤膜弃初滤液,取续滤液2 mL作为供试品溶液。
精密称定伊潘立酮对照品10 mg,置100 mL量瓶中,加流动相适量使溶解并稀释至刻度,摇匀。精密量取上述伊潘立酮对照品溶液1 mL置50 mL量瓶中,加溶出介质稀释至刻度,摇匀,作为对照品溶液。
将对照品溶液、供试品溶液分别注入液相色谱仪,记录色谱图,计算每片制剂各时间点的溶出量,绘制相应的溶出度曲线,取两条曲线上各时间点的平均溶出度值,采用非模型依赖法中的相似因子(f2)法计算相似因子。
相似因子计算公式如下:
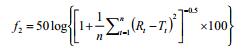
|
其中n为取样时间点个数,Rt为参比制剂的溶出度值,Tt为自研制剂的溶出度值[4]。
2.3 多溶媒溶出曲线测定结果伊潘立酮参比制剂与自研制剂在4种不同pH溶出介质中的平均溶出度曲线(n=12)见图 2。

|
图 2 伊潘立酮参比制剂(A)与自研制剂(B)在4种溶出介质下的溶出曲线 Fig. 2 Dissolution curves of reference formulation (A) and the self-prepared formulation (B) in four different media |
2.4 溶出一致性评价结果
伊潘立酮参比制剂与自研制剂在4种不同pH溶出介质中的相似因子拟合结果见表 3。由结果可知,在0.1 mol/L盐酸和pH4.5醋酸盐缓冲液中,自研制剂与参比制剂在15 min内的溶出量>85%,可判定为溶出行为相似,无需进行f2拟合[5];在pH6.8磷酸盐缓冲液和水中,选择5、10、15 min进行相似因子拟合,f2值均大于50,溶出行为相似。
| 表 3 多溶媒溶出相似因子拟合 Table 3 f2 factors between two tablets in four different media |
3 讨论
近年来研究认为,基于生物药剂学分类系统(Biopharmaceutical Classification System,BCS)理论的溶出试验是最能替代药物生物等效性研究的体外试验方法[6],当制剂的最大规格对应的原料药在250 mL(或更少)pH1.0~6.8的水溶性介质中自由溶解则可认为该药物是高溶解性药物[7]。伊潘立酮属于低溶解度高渗透性的BCS II类药物[8-9],研究表明,BCS Ⅱ类药物出现生物不等效的风险比Ⅰ、Ⅲ类药物高4倍[10],因此伊潘立酮片的体外溶出行为直接影响其体内生物利用度,需要通过与参比制剂溶出曲线的比较,来推测在生物利用度方面可能存在的差异。
伊潘立酮片作为精神类药物,为提高精神分裂症患者口服给药的顺应性,要求片剂能够迅速释放,即在500 mL 0.1 mol/L盐酸、pH 4.5缓冲介质、pH6.8缓冲介质中,50 r/min下,药物在30 min内的溶出量均应达到标示量的85%以上[7]。根据日本《仿制药生物等效性试验指导原则》,难溶性药物制剂通常选择pH1.2、pH3.0~5.0、pH6.8的缓冲溶液、水作为体外一致性的溶出介质[11]。综合“pH-溶解度”曲线结果及相关指导原则,本文选择了0.1 mol/L盐酸溶液、pH4.5缓冲溶液、pH6.8缓冲溶液、水4种溶出介质,同时满足了人体内消化道各器官的变化范围。结果表明,伊潘立酮自研制剂与参比制剂在上述4个pH溶出介质中都有一定的崩解、溶出,且溶出速度与“pH-溶解度”测定结果相一致:pH4.5醋酸盐缓冲溶液>0.1 mol/L盐酸>pH6.8磷酸盐缓冲溶液>水。
本研究选取了4种不同pH的溶出介质,对自研制剂和参比制剂进行了溶出度一致性评价,两种制剂在四种溶出介质中溶出曲线均具有相似性,表明自研制剂与参比制剂能够达到体外溶出一致,且理论上对任何人群均应有较高的生物利用度,为生物等效性试验等提供了可靠基础。
| [1] | 刘岩, 翟金国, 杨涛. 新型非典型抗精神病药伊潘立酮临床应用新进展[J]. 精神医学杂志, 2015,28(5):384–387. |
| [2] | 中国药典[S].四部. 2015.通则8004, 8006. |
| [3] | 普通口服固体制剂溶出度试验技术指导原则[S]. 2015 |
| [4] | 张启明, 谢沐风, 宁保明. 采用多条溶出曲线评价口服固体制剂的内在质量[J]. 中国医药工业杂志, 2009,40(12):946–950. |
| [5] | 谢沐风. 溶出曲线相似性的评价方法[J]. 中国医药工业杂志, 2009,40(04):308–311. |
| [6] | Amidon G L, Lennernäs H, Shah V P, et al. A theoretical basis for biopharmaceutic drug classification:the correlation of in vitro drug product dissolution and in vivo bioavailability[J]. Pharmaceut Res, 1995, 12(3):413–420. doi:10.1023/A:1016212804288 |
| [7] | FDA. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system guidance for industry.[S]. 2015 |
| [8] | Dickinson P A, Lee W W, Stott P W, et al. Clinical relevance of dissolution testing in quality by design[J]. The AAPS Journal, 2008, 10(2):280–290. |
| [9] | Arif S A, Mitchell M M. Iloperidone:A new drug for the treatment of schizophrenia[J]. Am J Health Syst Pharm, 2011, 68(4):301–308. doi:10.2146/ajhp100079 |
| [10] | Cristofoletti R, Chiann C, Dressman J B, et al. A comparative analysis of biopharmaceutics classification system and biopharmaceutics drug disposition classification system:a cross-sectional survey with 500 bioequivalence studies[J]. J Pharmaceut Sci, 2013, 102(9):3136–3144. doi:10.1002/jps.23515 |
| [11] | 仿制药生物等效性试验指导原则[S]. 2012. |