 2015, Vol. 38
2015, Vol. 38引用本文 | doi: 10.7501/j.issn.1674-6376.2015.01.004 |



2. 中国医科大学药学院, 辽宁 沈阳 110001;
3. 中国石油天然气集团公司中心医院 药学部, 河北 廊坊 065000;
4. 安捷伦科技(中国)有限公司, 北京 100000
2. College of Pharmacy, China Medical University, Shenyang 110001, China;
3. Department of Pharmacy, Central Hospital of China National Petroleum Corporation, Langfang 065000, China;
4. Agilent Technologies Co., Ltd., Beijing 100000, China
代谢组学被定义为定量分析与时间相关的由于生命体病态生理刺激或者基因改变后产生的代谢物,是系统生物学中自上而下研究方法的一个分支[1],正活跃于公共或者私人的研究机构中,尤其是在植物代谢组研究、药物开发以及临床医学等领 域[2,3,4,5,6,7,8,9]。其分析方法有GC-MS、(U)HPLC-MS、NMR、(U)HPLC-NMR-MS等多种手段[10,11,12]。其中NMR分析方法,由于对于样品分析的无损伤性和无偏向性成为代谢组学分析的重要手段。在应用这些分析方法对经实验设计的从动物或者植物中得到的生物样品进行分析后,得到的原始数据一般要经过数据预处理、模式识别、生物样品定性/定量、代谢异常途径指认等过程,才能达到基本研究目的。
在代谢组学数据预处理阶段,从图谱中得到的数据需经过积分、归一化、峰对齐等步骤,才能将由于不同仪器或者样品受外界影响甚至操作所产生的谱图差异减小到最低,以减少由于非实验设计因素所产生的差异对实验研究结果造成的影响。其中对结果影响最大的是积分步骤。积分是指将NMR或者LC(GC)-MS所得到的数据,根据一定的区间(如NMR一般为δ0.04)或者指定一定积分数目(如300个区间),将区间内的或者所有数据根据指定数目,将峰强度积分,积分后的个数作为下一阶段数据处理中的自变量个数,峰强度作为自变量的数值。这样做的目的是防止由于样品受到pH、测试时温度等外界影响对于仪器的响应位移所发生的改变,如NMR中某个物质由于pH的微小差异引起化学位移的偏移;GC-MS或者(U)HPLC-MS中保留时间的微小变化,减少变量数量便于统计分析。但是,如果样品间的差异过大,现有的积分方法就不会达到所期望的效果,单纯的增加区间范围或者减少积分个数会降低谱图的分辨率,将两个峰积分为一个整体,从而导致代谢物质定性缺失;而减少区间范围或者增多积分个数虽然提高了谱图分辨率,但是却没有起到积分的应用目的[13]。本文以NMR分析SD大鼠随年龄增长尿样中内源性代谢物的改变实验数据为基础,提出了新的积分方法。此方法是将积分区间在一定范围内根据峰的位置变动(如δ0.04±0.02)而改变,既能够不降低谱图的分辨率,又能够合理地将化学偏移影响减少到最低,完整地对信号峰进行积分,确保差异代谢物指认的准确性。
1 材料与方法 1.1 仪器与试剂1H-NMR样品测试仪器为Bruker DRX 600 NMR(Reinstetten,Germany),Norell标准核磁管(ST500—75 mm o.d.,Norell,Inc.,Landisville,NJ,USA),三(三甲基硅烷)磷酸酯(TMSP,98 atom% D,Cambridge Isotope Laboratories,Inc)作为NMR化学位移δ 0.0校正与内标试剂,重水(D2O,99.8 atom % D,Schweres Wefsser,Norell,Inc),磷酸一氢钠与磷酸二氢钠(分析纯,汕头陇西化工厂)。
1.2 动物实验与样品收集成熟雄性SD大鼠(购自沈阳药科大学动物实验中心,动物许可证号SYXK( 辽) 2003-0013)8只,初始体质量为200 g。在正式实验前动物适应实验室与代谢笼2周。实验动物在室温22±2 ℃,12 h昼夜交替,相对湿度为45%~65%条件下饲养。自由饮水与饮食。分别于第1周、第12周收集大鼠24 h尿样(4 ℃左右),记为“UZ”和“UB”。3 000 r/min离心10 min,分取上清液,于−80 ℃保存。
1.3 样品制备与数据采集取−80 ℃冷藏尿样,在室温(20 ℃)下自然融化,于13 000 r/min离心10 min后,上清液用0.45 μm滤膜滤过。取续滤液400 μL与重水磷酸缓冲液(PBS缓冲液调节pH=7.4,50 μL D2O,10 mmol/L TMSP)250 μL,混合。取600 μL涡旋后样品溶液转移到核磁管中。1H测试频率为600.129 MHz,298.16 K。水峰抑制脉冲应用Bruker “noesypr1d”脉冲序列(RD-90°-t1-90°-tm-90°-acquire FID),延时时间t1=3 s,混合时间tm=100 ms。每个样品扫描64次,数据采集点为131,072(128 K),谱图宽度为12 019.230 Hz,采集时间为2.73 s。
1.4 NMR数据模式识别
所有尿样1H核磁数据使用Topspin(version 2.1pl1,Bruker Biospin,Rheinstetten,Germany)手动进行基线与相位校正,化学位移校正TMSP于δ 0.0。化学位移区间δ 4.7~5.0、δ 5.5~6.0因水峰抑制和尿素峰的影响而去除。积分区间δ 0.2~4.7、δ 5.0~5.5和δ 6.0~10.0使用软件ProMetab Suite version 1.4,应用两种不同的积分方法进行积分:①以δ0.04为间距固定步长积分方法积分;②应用变步长积分(adaptive binning)方法进行积分。积分后数据由ProMetab Suite根据每个图谱峰总和进行归一化,导出为TXT文本。数据矩阵用Pareto scaling(1/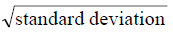 )方法做数据预处理。主成分分析(PCA)和偏最小二乘法-判别分析(PLS-DA)使用SIMCA-P软件包(version 11.5,UMETRICS AB,Sweden)进行分析。PCA分析用以可视化显示样品分类情况,溢出点判别,以及趋势判断,PLS-DA分析应用于聚类后生物标志物发现。
)方法做数据预处理。主成分分析(PCA)和偏最小二乘法-判别分析(PLS-DA)使用SIMCA-P软件包(version 11.5,UMETRICS AB,Sweden)进行分析。PCA分析用以可视化显示样品分类情况,溢出点判别,以及趋势判断,PLS-DA分析应用于聚类后生物标志物发现。
变步长积分方法包含以下步骤:①样本数据滤噪;②峰谷点辨识;③根据输入参数进行区间积分;④输出积分区间数值。
1.5.1 样本数据滤噪在没有信号的区间,如δ −1.0~3.0,计算此区间内噪音信号的均值与SD值,根据Bruker AMIX报道,图谱噪音noisylevel=mean(δ range)+noisyfactor×SD,其中noisyfactor一般为3.5~5.0。根据图谱噪音水平对图谱进行滤噪,选择高于噪音信号值的数据点为信号数据,或者采用小波分析的方法对全部谱图进行滤噪。
1.5.2 峰谷点辨识将所有谱图数据加和的均值作为标准谱图,用于寻找峰谷点。这样能够满足不同组分差异峰的峰谷点辨识,全面而又不丢失所有的峰数据。spectra_matrix_ref为输入标准图谱数值矩阵。对YS求两次差分,算法如下:
SlopeSign = diff (spectra_matrix_ref) > 0;
SlopeSignChange = diff (slopeSign) > 0;%过滤掉局部最低点微小的数据波动
h = find (slopeSignChange) + 1;%标记差分后最低点峰谷索引地址值
peakvalleyfind ( : ,2) = spectra_matrix(h);%输出峰谷点数值
1.5.3 变积分方法积分初始化参数,max_i_bin = bin_stepsize.×(1+mean_range);min_i_bin = bin_stepsize.×(1-mean_range),其中max_i_bin为设置区间中最大值(上边界),min_i_bin为设置区间中最小值(下边界),bin_stepsize为积分区间(一般为δ0.04),mean_range为区间变动百分比(一般为50%),matrix为计算得出峰谷点数值。
具体算法实现如下(Appendix I:MATLAB算法实现):
For峰谷点数值长度,将数值分段处理
res = 上段积分后残差与新数值段之和
If res > max_i_bin
将res根据bin_stepsize分段
res作为max_i_bin分段后残差
If res小于min_i_bin大于0
将res加入新数值矩阵段
res = 0
Elseif res大于min_i_bin,小于bin_stepsize
保存res值
End
Elseif res > = min_i_bin & res< = max_i_bin
res = 0;
Else % res<min_i_bin
保存res值
End
End
If res~ = 0 % res最后剩余值不大于最小值
新矩阵加入res
End
new_matrix = 新矩阵为分段区间边界点数值
NMR样品根据分段区间边界点,将区间内的峰强度积分。
1.5.4输出积分区间数值
2 结果与讨论 2.1 模式识别分析比较以δ0.04为区间固定步长积分方法和变步长积分方法积分后的数据经过PCA、PLS-DA方法进行模式识别分析,得到结果见表 1。
| 表 1 两种积分方法数据经模式识别分析的结果比较 Table 1Comparison on PCA and PLS-DA results between two methods |
表中结果表明,变步长积分方法在PCA、PLS-DA对于X(自变量)的解释能力(R2X值)要比固定步长积分方法更强,对于PLS-DA建立预测模型的预测能力(对因变量Y的解释能力,R2Y值)也要更好。这说明变步长积分方法能够有效地判断差异信号峰所引起的变化。
对于两种不同的积分方法采用PCA分析后计算R距离来考察其组内聚合、组间分离能力,其中R定义为R=Distance(Between Groups)/∑[Distance(Within Group)]。R值反映了样品聚类分散性的大小,R值越大说明其样品聚类性越好,也就是说组内间距小而组间间距大。通过比较计算后组内距离和组间距离以及R值可知,变步长积分方法能够有效的减少组内距离,增加组间距离,有效地将两个不同的类进行聚类(表 2)。
| 表 2 组内距离和组间距离以及R值 Table 2 Distances of samples within group and between groups and R value |
数据预处理的目的是减少主观因素引起的差异,放大试验设计所要考察的差异。数据预处理阶段所引入的不准确的差异数据会直接影响数据处理后的差异代谢物指认,导致由差异代谢物在代谢网络中的不全面而引起的解释错误。两种积分方法得到的潜在生物标志物是有所不同的(表 3),这种不同很可能是因为数据预处理阶段人为因素引入了误差。
| 表 3 两种积分方法PLS-DA中的VIP Table 3 VIP of PLS-DA between two methods |
下面以δ 4.008 9~3.96区间为例分析造成这种误差的原因。综合固定步长积分方法与变步长积分方法两方面,将这个区间分为两部分δ 4.008 9~3.984 9和δ 3.984 9~3.96。从图 1中可以看到,在固定步长积分方法中,δ 4.00~3.96为正相关(图 1A);而在变步长积分方法中δ 4.008 9~3.984 9为正相关,δ 3.984 9~3.964 4为负相关(图 1B)。从图 2中可以看出,在δ 4.008 9~3.96区间,“UB”组样品存在两个单峰(s),化学位移分别为δ 3.99,δ 3.97;“UZ”组样品在δ 3.975处存在一个双峰(dd)。固定步长积分方法不能够有效地区分这两组不同的峰的影响,而统一将其认为是一个弱的正相关(由于存在负相关的部分抵消,正相关性减弱);变步长积分方法却能够有效地发现这两组差异峰的存在,正确地判断相关性,从而减少由于数据预处理所引起的潜在生物标志物丢失,减少了由于正负相关性的抵消所产生的相关性值的减小,以及使显著的潜在生物标志物成为非显著生物标志物的现象。
 | 图 1 固定步长(A)和变步长(B)积分方法的载荷图 Fig.1 Loadingplotsof normal binning method (A) and adaptive binning method (B) |
 | 图 2 在δ 4.008 9~3.96两组样品谱图的区别 Fig.2 Differences between two spectra from two different groups at region δ 4.008 9—3.96 |
 | 图 3 固定步长积分(步长δ0.04)和变步长积分(δ0.04 ± 0.02)方法在δ7.68处的边界Fig.3 Marked borders of normal binning method (fix range δ0.04) and adaptive binning method (δ0.04 ± 0.02) at region δ7.68 |
由于固定步长积分方式没有考虑到峰位置,所以会将差异峰分裂为两部分,减弱差异峰对于分组影响的权重。以δ 7.68处单峰(s)为例(图 3)。δ7.68处单峰在固定步长积分方法中被分为两部分分别积分,而变步长积分方法能够识别峰的位置将信号峰作为一个整体进行积分。PCA载荷图中显示了这个差异对数据分析的影响(图 4)。
 | 图 4 固定步长(A)和可变步长(B)积分方法载荷图Fig.4 Loading plots of normal binning method (A) and adaptive binning method (B) |
综上分析,变步长积分方法能够有效地根据信号峰的位置对其进行积分,减少了不同信号峰被合并积分或者同一信号峰被拆分积分的情况。由于这一特点,变步长积分方法能够有效地增强判断样品聚类能力同时减少由于积分方法产生的差异代谢物丢失,解决固定步长积分方法不能够同时兼顾图谱分辨率和减少由于环境引起的化学位移变化的矛盾。
变步长积分方法也可以扩展应用到三维数据中(如GC-MS、HPLC-MS采集的数据),增强数据预处理的可靠性,避免由于误差引起的差异代谢物、生物标志物指认错误。
致谢:感谢李发美实验室各位老师和研究生郑姝宁、唐静同学在实验过程中给予的帮助。
| [1] | Keun H C. Metabonomic modeling of drug toxicity[J]. Pharmacol The, 2006, 109(1-2): 92-106. |
| [2] | Schripsema J. Application of NMR in plant metabolomics: techniques, problems and prospects[J]. Phytochem Anal, 2010, 21(1): 14-21. |
| [3] | Ala-Korpela M. Potential role of body fluid 1H NMR metabonomics as a prognostic and diagnostic tool[J]. Expert Rev. Mol Diagn, 2007, 7(6): 761-773. |
| [4] | Aich P, Babiuk L A, Potter A A, et al. Biomarkers for prediction of bovine respiratory disease outcome[J]. OMICS, 2009, 13(3): 199-209. |
| [5] | Ala-Korpela M. Critical evaluation of 1H NMR metabonomics of serum as a methodology for disease risk assessment and diagnostics[J]. Clin Chem Lab Med, 2008, 46(1): 27-42. |
| [6] | Lindon J C, Holmes E, Nicholson J K. Metabonomics in pharmaceutical R&D[J]. FEBS J, 2007, 274(5): 1140-1151. |
| [7] | Coen M, Holmes E, Lindon J C, et al. NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology[J]. Chem Res Toxicol, 2008, 21(1): 9-27. |
| [8] | Duarte I F, Diaz S O, Gil A M. NMR metabolomics of human blood and urine in disease research[J]. J Pharm Biomed Anal, 2014, 93C: 17-26. |
| [9] | Beger R D, Sun J, Schnackenberg L K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity[J]. Toxicol Appl Pharmacol, 2010, 243(2): 154-166. |
| [10] | Zhang M, Deng M, Ma J, et al. An evaluation of acute hydrogen sulfide poisoning in rats through serum metabolomics based on gas chromatography-mass spectrometry[J]. Chem Pharm Bull, 2014, 62(6): 505-507. |
| [11] | Lee D K, Lim D K, Um J A, et al. Evaluation of four different analytical tools to determine the regional origin of Gastrodia elata and Rehmannia glutinosa on the basis of metabolomics study[J]. Mol, 2014, 19(5): 6294-6308. |
| [12] | Sarker S D, Nahar L. Hyphenated techniques and their applications in natural products analysis[J]. Methods Mol Biol, 2012, 864: 301-340. |
| [13] | Jacob D, Deborde C, Moing A. An efficient spectra processing method for metabolite identification from 1H-NMR metabolomics data[J]. Anal Bioanal Chem, 2013, 405(15): 5049-5061. |